X-ray, Elektron dan Penghabluran NMR kepada Penentuan Struktur Molekul Organik Kecil
BERITA JEOL Jld.53 No.6
Yusuke Nishiyama1, 2
1 JEOL RESONANCE Inc. 2 Pusat Kerjasama RIKEN CLST-JEOL
Di sini kami membentangkan perkembangan terkini di makmal kerjasama RIKEN CLST-JEOL untuk meneroka struktur molekul sebatian farmaseutikal berat molekul rendah dalam kelimpahan semula jadi (tanpa sebarang pelabelan isotop); kemajuan terkini dalam teknologi pemutar sudut ajaib (MAS) pantas dalam resonans magnetik nuklear keadaan pepejal (ssNMR) dan dalam kamera kepekaan ultra tinggi dalam mikroskop elektron penghantaran (TEM) membuka cara baharu untuk menjawab masalah dalam industri dan sains farmaseutikal. 1) Polimorf kristal dan 2) garam/kokristal adalah dua kebimbangan utama dari segi kawalan kualiti, kestabilan dan harta intelek. Untuk mengenal pasti bentuk kristal, pembelauan sinar-X serbuk dan 13C pengutuban silang MAS ssNMR adalah kaedah yang digunakan secara meluas, walau bagaimanapun, yang pertama kadangkala tidak sesuai untuk analisis campuran dan yang kedua gagal membezakan bentuk kristal dengan konformasi molekul yang serupa. Untuk menyelesaikan isu ini, kami menggunakan pembelauan elektron (ED) dan 1H cepat MAS NMR. Bentuk hablur boleh ditentukan daripada hablur tunggal bersaiz nano hingga mikro meter menggunakan ED, kerana interaksi elektron adalah 4 hingga 5 tertib lebih kuat daripada interaksi sinar-X. 1H NMR juga memberikan maklumat yang sesuai kepada pembungkusan molekul sejak itu 1H terletak pada permukaan hablur. Isu garam/kokristal, di mana hidrogen memainkan peranan penting, adalah masalah yang serius, kerana pembelauan sinar-X kristal tunggal (SCXRD) tidak dapat menentukan kedudukan atom hidrogen dengan tepat. Di sini kita menentukan jarak antara nuklear 1H dan 15N menggunakan ssNMR pada keadaan MAS yang pantas, manakala struktur global diperoleh melalui SCXRD, menjawab soalan garam/kokristal.
Pengenalan
Walaupun ubat berasaskan bioteknologi disenaraikan di bahagian teratas dalam pasaran farmaseutikal, ubat tradisional berat molekul rendah masih sangat penting untuk rawatan harian, contohnya penyakit dewasa. Bahan farmaseutikal aktif (API) berat molekul rendah ini biasanya boleh dihablurkan dalam beberapa bentuk berbeza, iaitu polimorf hablur, bergantung pada keadaan penghabluran. Memandangkan keterlarutan dan kestabilan sebahagian besarnya dipengaruhi oleh bentuk kristal, adalah sangat penting untuk mengawal dan memantau bentuk kristal dari sudut pandangan kawalan kualiti dari peringkat pembangunan hingga pengeluaran [1, 2, 3]. Apabila kristal dengan saiz yang cukup besar (~100 μm) tersedia, pembelauan sinar-X kristal tunggal (SCXRD) memberikan jawapan yang berbeza kepada bentuk kristal dengan resolusi atom. Walau bagaimanapun, kebanyakan ubat berat molekul rendah disediakan dalam bentuk kristal mikro dengan pelbagai formulasi termasuk tablet, pil, serbuk. Oleh kerana formulasi ini melibatkan bahan bantu, adalah penting untuk menentukan bentuk kristal daripada kristal mikro dalam campuran. Belauan sinar-X serbuk (PXRD) dan 13C silang polarisasi sudut ajaib berputar resonans magnet nuklear (CPMAS NMR) adalah dua kaedah utama untuk mengenal pasti bentuk kristal. Oleh kerana corak/spektra eksperimen memberikan cap jari bagi setiap bentuk kristal, perbandingan corak/spektra antara ubat dan bentuk piawai memberikan, dalam kebanyakan kes, jawapan yang jelas kepada kristal dari dalamnya. Walau bagaimanapun, kedua-dua kaedah masih mempunyai masalah praktikal. PXRD kadangkala gagal untuk mengenal pasti bentuk kristal kerana banyak patters pembelauan daripada API dan eksipien bertindih antara satu sama lain. Selain itu, 13C CPMAS yang sensitif kepada konformasi molekul adalah kaedah yang sesuai untuk analisis campuran kerana kebanyakan isyarat daripada eksipien muncul pada kedudukan yang berbeza daripada API, mengelakkan pertindihan isyarat. Walau bagaimanapun, 13C CPMAS agak tidak sensitif kepada pembungkusan molekul kerana atom karbon tertimbus di dalam molekul dan terletak jauh dari permukaan molekul. Oleh itu, 13C CPMAS gagal mengenal pasti bentuk kristal dengan konformasi molekul yang serupa. Selain itu, sensitiviti kepada bentuk kristal adalah kurang daripada PXRD.
Isu utama lain dalam API berat molekul rendah ialah cara untuk mengenal pasti garam/kokristal/kontinum dalam sistem berbilang komponen. Kadangkala diperlukan untuk meningkatkan keterlarutan dan/atau kestabilan kristal API. Sistem berbilang komponen yang terdiri dengan API dan koformer lengai merupakan salah satu penyelesaian yang digunakan secara meluas untuk isu ini. Banyak contoh boleh didapati dalam sistem berbilang komponen dengan mencampurkan API (biasanya asas) dan koformer (berasid). Apabila perbezaan pKa (ΔpKa) lebih besar daripada 3, ini membentuk garam, di mana interaksi ionik antara molekul ditemui. Dalam garam, proton dari coformer bergerak sepenuhnya ke arah API. Baru-baru ini, satu lagi kelas sistem berbilang komponen, kokristal, diperkenalkan apabila ΔpKa kurang daripada 3. Dalam kohablur, proton dalam koformer masih kekal di sana dan ikatan hidrogen antara molekul terbentuk. Selain itu, sistem antara garam dan kokristal juga ditemui iaitu kontinum di mana proton terletak di antara API dan koformer. Adalah penting untuk mengenal pasti garam/kokristal/kontinum dari segi sudut pandangan intelek, terutamanya apabila ΔpKa lebih kecil daripada 3. Walau bagaimanapun, kekurangan keupayaan untuk menentukan kedudukan proton dalam kaedah berasaskan XRD membawa isu serius kepada pengenalpastian garam/kokristal/ kontinum, kerana perbezaan antara mereka hanya datang dari kedudukan hidrogen.
Isu utama yang belum diselesaikan ialah 1) analisis campuran, 2) pengenalpastian bentuk kristal dengan bentuk molekul yang serupa, dan 3) kekurangan keupayaan untuk menentukan 1jawatan H. Dua isu terdahulu berkaitan dengan polimorf kristal dan yang terakhir ialah garam/kokristal/kontinum. Di sini, kami menggabungkan pembelauan elektron (ED), 1H ssNMR, dan SCXRD untuk menangani isu ini. ED adalah salah satu kaedah pemerhatian dalam peralatan mikroskop elektron penghantaran (TEM) dan memberikan corak pembelauan. Oleh kerana interaksi elektron adalah 104 hingga 105 lebih kuat daripada sinar-X, corak ED boleh diperhatikan daripada kristal tunggal bersaiz nano kepada mikro, membolehkan analisis campuran. Walaupun penggunaan ED sebahagian besarnya terhad kepada bahan bukan organik yang teguh kepada penyinaran elektron, kemajuan terkini dalam kamera kepekaan tinggi membolehkan mod dos rendah bersama dengan pemegang sampel cryo, membuka jalan kepada pemerhatian ED bagi kristal organik termasuk molekul rendah. API berat. Isu 2) dibangkitkan kerana 13C CPMAS tidak sensitif kepada konformasi molekul seperti yang dinyatakan di atas. Mujurlah, pengenalan teknologi MAS yang sangat pantas > 70 kHz membolehkan resolusi tinggi 1Pemerhatian H walaupun dalam pepejal tegar [4, 5, 6], di mana 1Hs berganding rapat dengan 1H-1Interaksi dipolar H pada kadar MAS sederhana. Sejak 1H terletak di permukaan, 1Anjakan kimia isotropik H sensitif kepada bukan sahaja konformasi tetapi juga pembungkusan molekul. Sebagai tambahan, 1H-1Korelasi antara molekul H memberikan corak yang berbeza untuk setiap pembungkusan molekul. Teknologi MAS yang pantas merealisasikan bukan sahaja resolusi tinggi 1H NMR tetapi juga beberapa eksperimen yang canggih, termasuk 14N NMR dan 1H-15N ukuran jarak. Yang pertama mendedahkan keadaan terprotonasi dalam molekul organik kecil. Yang terakhir memberikan jawapan yang jelas kepada isu garam/kokristal/kontinum di mana kedudukan hidrogen memainkan peranan penting. Di samping itu, kami menilai daya pengeluaran setiap kaedah.
Polimorf Kristal[7]
Pengenalpastian bentuk kristal dalam aplikasi farmaseutikal adalah penting dan biasanya dilakukan oleh PXRD dan 13C CPMAS ssNMR. Walau bagaimanapun, yang pertama tidak sesuai untuk analisis campuran dan yang terakhir kadang-kadang gagal seperti yang dibincangkan di atas. Sebagai contoh, 13C Spektrum CPMAS bagi tiga polimorf L-histidin (pseudo) berbeza ditunjukkan dalam Rajah 1. L-histidin (Rajah 1b, c) memberikan spektrum yang berbeza daripada garam hidroklorida L-histidin·HCl·H2O (Rajah 1a), kerana konformasi molekul yang berbeza. Sebaliknya, L-histidine dalam bentuk ortorombik (Rajah 1b) dan monoklinik (Rajah 1c) memberikan hampir sama. 13C spektrum CPMAS, mencerminkan konformasi rapat antara satu sama lain. Seperti yang ditunjukkan di sini, 13C CPMAS adalah ukuran sensitif untuk mengenal pasti bentuk kristal dengan konformasi yang berbeza, namun, gagal untuk membezakan polimorf dengan konformasi molekul yang serupa. Kami di sini mencadangkan pendekatan gabungan ED dan 1H ssNMR di MAS yang sangat pantas untuk menjawab soalan-soalan ini.
Rajah 2 menunjukkan imej TEM dan corak ED L-histidine·HCl·H2O diukur pada suhu bilik. Walaupun saiz kristal adalah kira-kira 1 μm, yang biasanya terdapat dalam produk farmaseutikal, 100 nm nano-beam digunakan. Oleh itu, pada dasarnya, kristal dengan 100 nm cukup besar untuk mendapatkan corak ED kristal tunggal. Malah, kami telah mengukur corak ED dari 100 nm atau bahkan kristal yang lebih kecil. Sebagai dos kritikal L-histidine·HCl·H2O pada suhu bilik hanya 10-20 e-nm-2, kami menetapkan kadar dos kepada 10 e-nm-2s-1 dengan masa pendedahan 1 s, menghasilkan jumlah dos sebanyak 10 e-nm-2. Walaupun dengan keadaan dos yang sangat rendah, kamera CMOS terkini (OneView, Gatan, Inc., Amerika Syarikat) memberikan sensitiviti yang mencukupi. Manakala dos kritikal 10-20 e-nm-2 biasanya terdapat dalam molekul organik, pemegang sampel cryo menambah baik dos kritikal lagi jika diperlukan. Oleh kerana corak ED boleh dikira dengan mudah daripada struktur kristal, bentuk kristal boleh dikenal pasti dengan membandingkan corak ED eksperimen dan dikira. Corak ED L-histidine dalam bentuk ortorombik dan monoklinik ditunjukkan dalam Rajah 3 dan Rajah 4, masing-masing. manakala 13C CPMAS gagal membezakan kedua-dua bentuk ini (Rajah 1), pembelauan ED memberikan corak yang berbeza untuk setiap bentuk kristal. Ini kerana parameter kekisi adalah berbeza sama sekali dalam kedua-dua bentuk ini. Selain itu, ED mendayakan pengukuran daya pemprosesan yang tinggi dalam masa satu minit berbanding PXRD (berpuluh minit) dan 13C CPMAS (jam). Pengukuran ED dilakukan menggunakan JEM-ARM200F (JEOL Ltd., Jepun). Kebanyakan instrumen TEM mampu melakukan pengukuran ini jika kamera sensitiviti tinggi dipasang.
Pembungkusan molekul boleh disiasat menggunakan 1H NMR di MAS yang sangat pantas. 1D 1Spektrum H NMR polimorf L-histidin (pseudo) pada 70 kHz MAS ditunjukkan dalam Rajah 5, memberikan corak yang berbeza. Terutama, L-histidin dalam bentuk ortorombik (b) memberikan corak yang berbeza daripada satu dalam bentuk monoklinik (c). Perbezaan ini datang daripada pembungkusan molekul yang berbeza. Ini menyerlahkan kelebihan 1H NMR ke atas 13C CPMAS yang gagal mengenal pasti dua bentuk kristal ini. Terima kasih kepada kelimpahan yang tinggi (>99%) dan frekuensi Larmor (600 MHz pada 14.1 T), ia mengambil masa kurang daripada satu minit untuk memerhatikan spektrum 1D ini. Perbezaan antara dua bentuk kristal boleh diperbesarkan dengan memerhati 1H/1Eksperimen korelasi homonuklear H, yang sepatutnya sensitif kepada antara molekul 1H/1Kesambungan H, dengan itu pembungkusan kristal. Keupayaan korelasi homonuklear adalah salah satu ciri unik 1H NMR dengan bantuan sensitiviti dan kelimpahannya yang tinggi. Rajah 6 memberikan 2D 1H spektrum korelasi homonuklear kuantum ganda (DQ) / kuantum tunggal (SQ) L-histidin dalam bentuk ortorombik dan monoklin. Walaupun spektrum 1D hanya menunjukkan maklumat tempatan, spektrum korelasi homonuklear 2D juga mencerminkan maklumat tentang pembungkusan molekul, seperti yang ditunjukkan dengan jelas dalam spektrum bertindih. Memandangkan koheren DQ dicipta melalui 1H-1Interaksi dipolar H, keamatan puncak silang mencerminkan kedekatan spatial. Sebagai contoh, jarak internuklear antara H1 dan H4 ialah 4.1 Å untuk ortorombik, ia jauh lebih pendek dan 2.86 Å dalam bentuk monoklinik. Walaupun perbezaan jarak hanya 1.4 kali, ini menghasilkan interaksi dipolar 2.9 kali lebih kuat dalam bentuk monoklinik. Oleh itu, korelasi H1-H4 hanya diperhatikan dalam bentuk monoklinik. Ia adalah percubaan 2D, walau bagaimanapun, masa pengukuran biasanya kurang daripada satu jam, memberikan daya pemprosesan yang lebih tinggi daripada 13C CPMAS.
1H/14N korelasi NMR juga memberikan maklumat yang bermanfaat. Terdapat berbilang keadaan terprotonasi dalam L-histidine. Sama ada atau kedua-dua daripada dua nitrogen atτ dan δ pada cincin imidazol boleh diprotonasi kepada bahagian NH. Di samping itu, L-histidine boleh menjadi Zwitterion. Adalah mencabar untuk membezakan keadaan terprotonasi ini dengan sama ada XRD atau ED, kerana kedua-dua kaedah tidak sensitif kepada kedudukan hidrogen. Walau bagaimanapun, 1H/14Spektrum korelasi N L-histidin (Rajah 7) memberikan jawapan yang jelas. Dalam persekitaran berasid dalam L-histidine·HCl·H2O, tiga korelasi NH muncul. Ini jelas menunjukkan kedua-dua τ dan δ nitrogen terprotonasi. Satu lagi korelasi NH muncul pada kedudukan frekuensi rendah dalam 14Dimensi N pada -260 ppm mencadangkan bahawa asid amino ini adalah Zwitterion dengan NH3+ bahagian. Ini adalah kerana gandingan quadrupolar kecil dalam NH3+ disebabkan oleh simetri tempatan. Sebaliknya, dua polimorf L-histidin hanya memberikan dua puncak NH untuk setiap satu. Jelas sekali kedua-duanya adalah Zwitterion dan hanya satu daripada τ dan δ nitrogen terprotonasi. Perlu diingatkan bahawa 1H/14N biasanya mengambil masa kurang daripada 10 minit kerana kedua-duanya 1H dan 14N ialah nukleus yang banyak.
Semua ukuran di atas dilakukan menggunakan spektrometer JNM-ECZ600R (JEOL RESONANCE Inc., Jepun) dilengkapi dengan probe MAS ssNMR 1 mm pantas (JEOL RESONANCE Inc.) pada 14.1 T. Jumlah sampel yang digunakan dalam pengukuran adalah kira-kira 1 mg untuk setiap .
Fig.1
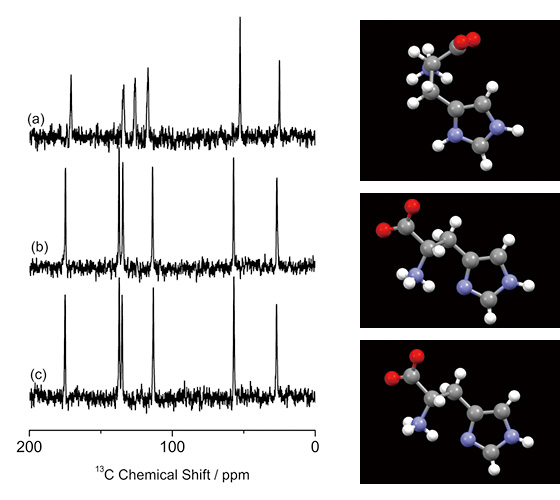
13C Spektrum CPMAS (a) L-histidin·HCl·H2O, (b) L-histidin (ortorombik) dan (c) L-histidin (monoklinik), bersama-sama dengan konformasi molekul. 256 imbasan untuk (a) dan 512 imbasan untuk (b) dan (c) telah terkumpul. (a) diukur pada 16.4 T dengan spektrometer JNM-ECA700II (JEOL RESONANCE Inc., Jepun). Angka itu diterbitkan semula daripada rujukan 7.
Fig.2

(a) imej TEM dan (b) pancaran nano (diameter 100 nm) corak difraksi mikrohabluran L-histidin·HCl·H2Wahai sampel. Corak pembelauan diperoleh daripada kawasan yang ditunjukkan oleh bulatan putih dalam (a). Angka itu diterbitkan semula daripada rujukan 7.
Fig.3
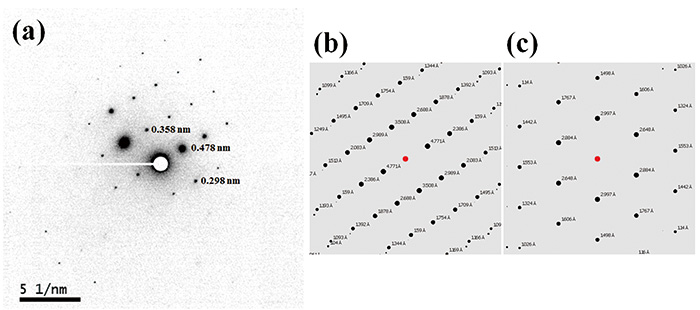
(a) Corak ED eksperimen yang dipaparkan secara negatif untuk L-histidine (orthorhombic) dan corak ED yang dikira untuk (b) L-histidine (orthorhombic) dan (c) L-histidine (monoklinik). Jarak-d yang sepadan dengan titik pembelauan dianggarkan oleh panjang kamera yang ditentukur 40 cm dan panjang gelombang 2.51 petang pada voltan pecutan 200 kV. Corak ED dikira untuk rasuk kejadian paksi zon [631]. Angka itu diterbitkan semula daripada rujukan 7.
Fig.4

(a) Corak ED eksperimen yang dipaparkan secara negatif untuk L-histidine (monoklinik) dan pola ED yang dikira untuk (b) L-histidine (ortorombik) dan (c) L-histidine (monoklinik). Jarak-d yang sepadan dengan titik pembelauan dianggarkan oleh panjang kamera yang ditentukur 40 cm dan panjang gelombang 2.51 petang pada voltan pecutan 200 kV. Corak ED dikira untuk rasuk kejadian paksi zon [100]. Angka itu diterbitkan semula daripada rujukan 7.
Fig.5
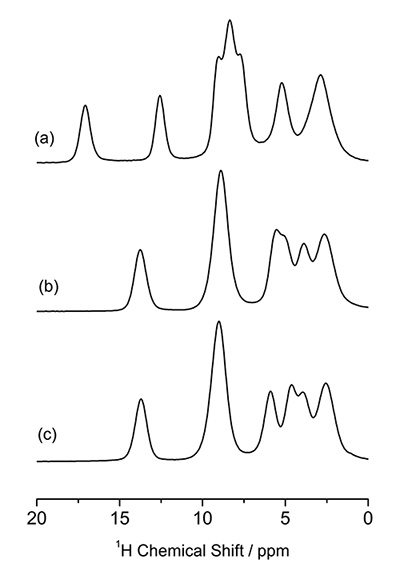
. 1Spektrum H NMR bagi (a) L-histidin·HCl·H2O, (b) L-histidin (ortorombik) dan (c) L-histidin (monoklinik). Angka itu diterbitkan semula daripada rujukan 7.
Fig.6
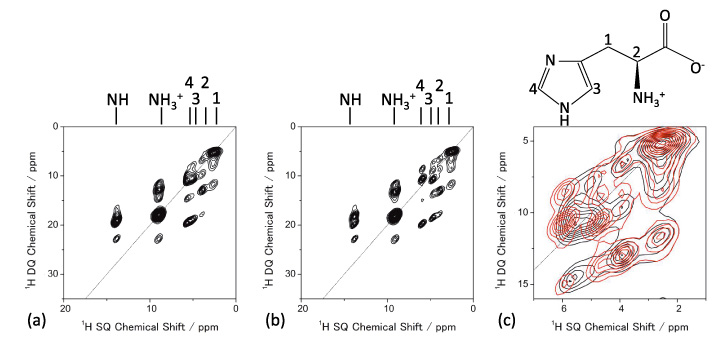
1H DQ/1Spektrum H SQ NMR bagi (a) L-histidin (ortorombik) dan (b) L-histidin (monoklinik). Pengembangan spektrum bertindih juga ditunjukkan dalam (c) (L-histidin (ortorombik) dalam warna hitam dan L-histidin (monoklinik) dalam warna merah). Empat imbasan untuk setiap t1 tempoh terkumpul dengan 32 t1 kenaikan. Angka itu diterbitkan semula daripada rujukan 7.
Fig.7
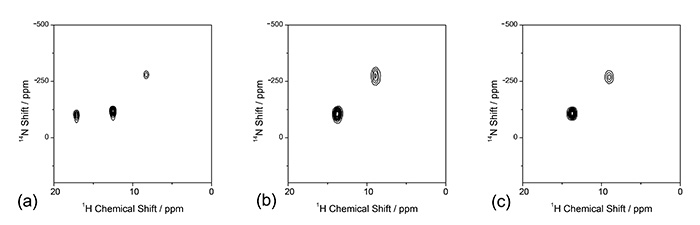
. 1H/{14N} Spektrum NMR bagi (a) L-histidin·HCl·H2O, (b) L-histidin (ortorombik) dan (c) L-histidin (monoklinik). Spektrum (a) diukur pada kadar 90 kHz MAS dengan probe HXMAS 0.75 mm (JEOL RESONANCE Inc., Jepun). Lapan imbasan untuk setiap t1 tempoh terkumpul dengan (a) 64 dan (b)/(c) 32 t1 kenaikan. Tidak 1H–14N gandingan semula digunakan semasa masa mencampurkan. Angka itu diterbitkan semula daripada rujukan 7.
Kaedah Baharu Dibangunkan untuk Polimorf Kristal
Walaupun kepentingan nitrogen dalam kimia, farmasi, sains bahan dan lain-lain, nitrogen NMR agak terhad. Ini adalah semata-mata kerana sensitiviti rendah 15N NMR kerana kelimpahan yang rendah 15N (0.4%). Walaupun 15N diutamakan kerana nombor kuantum putarannya I = 1/2, isotop lain bagi 14N juga merupakan nukleus aktif NMR dengan kelimpahan tinggi yang menggalakkan iaitu 99%. Nombor kuantum putaran integer (I = 1) dan kehadiran gandingan quadrupolar menghalang penggunaan 14N NMR. Untuk mengatasi masalah ini, kami membangunkan 1H-{14N} pengukuran koheren berbilang kuantum heteronuklear (HMQC) pada keadaan MAS yang sangat pantas [8]. Kaedah ini membolehkan pengukuran daya pemprosesan tinggi dengan sampel terhad jisim kurang daripada 1 mg. Kerana kepekaan yang tinggi, kaedah ini boleh dikembangkan kepada tiga dimensi untuk 14N/1H DQ/1Eksperimen korelasi H SQ[9]. Di samping itu, kaedah meneroka pemerhatian 35Cl yang sering terdapat dalam garam farmaseutikal [10].
Tugasan isyarat membantu mentafsir spektrum NMR seperti yang dibincangkan di atas dalam Rajah 6. Walaupun korelasi homo/heteronuklear dua dimensi banyak membantu untuk menetapkan isyarat melalui sambungan dua putaran, eksperimen tiga dimensi memberikan tugasan yang lebih jelas. Untuk tujuan ini, kami membangunkan 13C/1H DQ/1H SQ korelasi untuk mendedahkan tempatan 1H-1Rangkaian putaran H di sekitar 13C [11]. Tugasan lengkap dicapai daripada spektrum tiga dimensi tunggal dengan sampel 1 mg pada kelimpahan semula jadi.
Garam/Cocrystal/Continuum[12]
Seperti yang dibincangkan di atas, pengenalpastian garam/kokristal/kontinum masih kekal sebagai isu besar dalam aplikasi farmaseutikal kerana kekurangan keupayaan untuk mencari hidrogen dalam XRD. Perbezaan antara ketiga-tiga kelas ini boleh ditafsirkan sebagai jarak yang berbeza antara hidrogen dan nitrogen (Rajah 8). Oleh itu, saiz 1H-15Interaksi dipolar N, yang berkadar songsang dengan kubus jarak antara nuklear, harus memberikan jawapan yang jelas kepada garam/kokristal/kontinum.
Di sini, kami mencadangkan kaedah gabungan SCXRD dan ssNMR. Walaupun SCXRD menentukan struktur kristal global, ssNMR memberikan jarak internuklear tempatan. Dengan menggabungkan kedua-dua jenis maklumat ini, pemahaman yang lancar tentang struktur kristal dapat diwujudkan. Sebagai demonstrasi, pertama, kami mensintesis empat model sistem berbilang komponen yang ditunjukkan dalam Rajah 9. Struktur kristal ditentukan oleh SCXRD. Keputusan jelas menunjukkan kehadiran hubungan antara molekul antara N dan OH seperti yang dijangkakan (Rajah 10). Persoalannya adakah ia garam/kokristal/kontinum. Untuk menjawab soalan ini, kami cuba menentukan kedudukan hidrogen oleh SCXRD, namun, ia memberikan nilai bergantung kepada mesin, mengakibatkan kedudukan yang tidak boleh dipercayai. Oleh kerana struktur kristal yang ditentukan oleh SCXRD jelas menunjukkan, terdapat gandingan 1H/15N antara molekul. The 1H/15N jarak, sekali gus menggabungkan kekuatan antara 1H dan 15N, harus memberi jawapan kepada masalah garam/kokristal/kontinum. Sebagai contoh, 1H/15N jarak dalam SA2 ditentukan sebagai 1.25 Å (Rajah 10). Dengan jarak antara nitrogen dan oksigen 2.54 Å, yang ditentukan oleh SCXRD, kita boleh membuat kesimpulan SA2 ialah kontinum kerana hidrogen terletak di tengah-tengah oksigen dan nitrogen. Perlu diingatkan bahawa SA2 terdiri daripada asid dan bes kuat dengan ΔpKa lebih besar daripada 3. Keputusan ini menyerlahkan kepentingan pengukuran SCXRD/ssNMR untuk mengenal pasti garam/kokristal/kontinum walaupun untuk sistem dengan ΔpKa yang besar. Struktur dan jarak internuklear bagi tiga sistem yang lain telah berjaya ditentukan (Rajah 10). Kesesakan kaedah ini adalah masa percubaan untuk 1H-15N ukuran jarak. Di samping kelimpahan semula jadi yang rendah 15N dan kecil 1H-15N gandingan, sampel farmaseutikal ini biasanya menunjukkan sangat panjang 1HT1 masa bersantai. Selain itu, keperluan MAS yang pantas mengurangkan jumlah sampel, mengakibatkan pengurangan sensitiviti selanjutnya. Kami telah membangunkan kaedah untuk meningkatkan daya pengeluaran seperti yang dibincangkan dalam bahagian berikut, setiap pengukuran masih memerlukan 4-5 hari. Kami bersetuju dengan fakta pengeluaran terhad. Namun begitu, kami percaya kaedah ini agak berguna kerana tiada kaedah lain memberikan jawapan yang jelas.
Kesemua pengukuran NMR telah dijalankan dengan spektrometer NMR JNM-ECA700II (JEOL RESONANCE Inc.) menggunakan probe MAS cepat resonans berganda 1 mm (JEOL RESONANCE Inc.) pada 16.4 T.
Fig.8
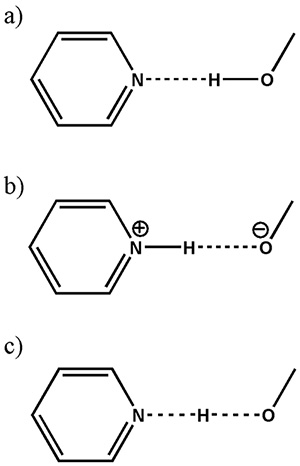
Persembahan skematik (a) kokristal, (b) garam dan (c) kontinum (di mana kedudukan atom H dikongsi antara dua atom berat) dalam O tipikal...H...N interaksi. Angka itu diterbitkan semula daripada rujukan 12.
Fig.9
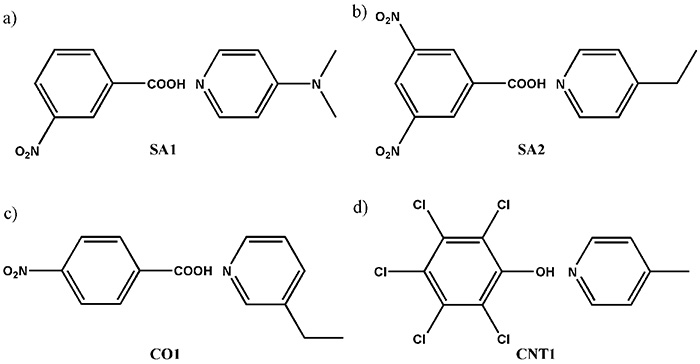
Perwakilan skematik bagi sebatian yang digunakan dalam kajian ini, menunjukkan (a) SA1 (asid 3-nitrobenzoik dan N,N-dimethypyridin-4-amine), (b) SA2 (asid 3,5-dinitrobenzoik dan 4-etilpiridin), (c) CO1 (asid 4-nitrobenzoik dan 3-etilpiridin) dan (d) CNT1 (pentachlorophenol dan 4-methylpyridine). Angka itu diterbitkan semula daripada rujukan 12.
Fig.10
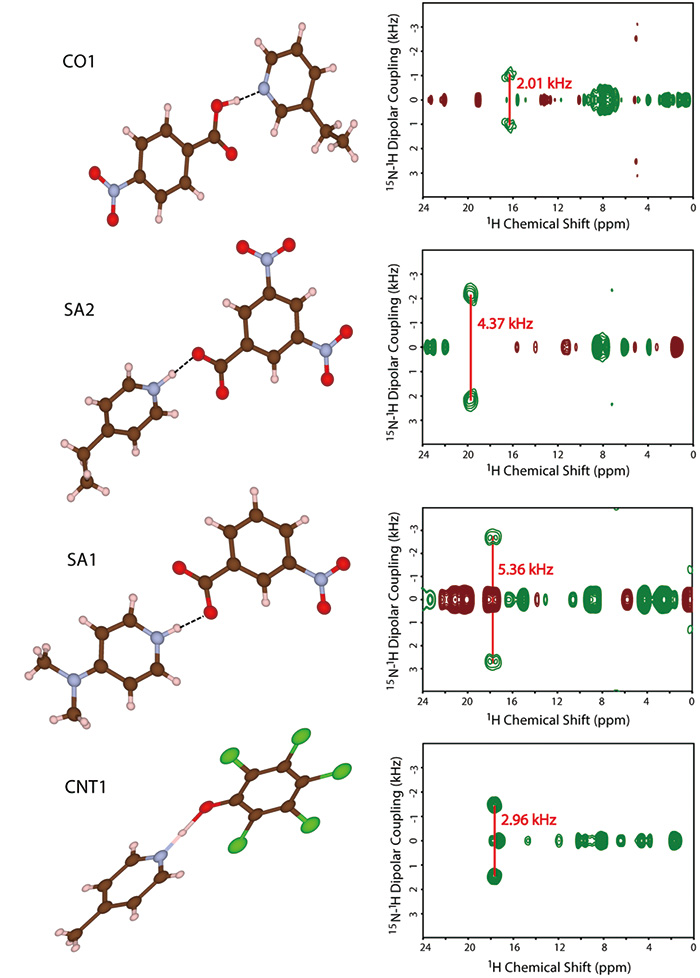
Struktur molekul dan spektrum invCP-VC dua dimensi (15N-1H gandingan dipolar berbanding 1anjakan kimia H) SA1, SA2, CO1 dan CNT1. Angka itu diterbitkan semula daripada rujukan 12.
Kaedah Baru Dibangunkan untuk Garam/Cocrystal/Continuum
Saiz 1H/15N gandingan dipolar hendaklah diukur dengan tepat. Walau bagaimanapun, ia bukan tugas yang mudah kerana 1) kelimpahan yang rendah 15N (0.4%), 2) nisbah gyromagnetik kecil (1/10 daripada 1H), dan 3) banyak 1Nukleus H yang tidak terikat kepada 15N, menghasilkan kepekaan rendah, gandingan dipolar kecil (1-7 kHz), dan pertindihan 1H resonans, masing-masing. Lebih penting lagi, kaedah sebelumnya cenderung untuk menunjukkan 1H/15N gandingan dipolar yang bergantung kepada keadaan eksperimen, memberikan jarak yang tidak boleh dipercayai. Untuk mengatasi kesukaran ini, kami telah memperkenalkan CP dikesan songsang dengan kaedah masa sentuhan berubah (inv CP-VC) pada MAS pantas [13]. Pertama, kaedah ini memberikan pemisahan dipolar yang jelas yang bebas kepada keadaan eksperimen [14]. Sebenarnya, faktor penskalaan kaedah inv CP-VC bergantung pada kekuatan medan rf. Walau bagaimanapun, isyarat dari kawasan dengan B1 ketidakhomogenan bertaburan pada julat spektrum yang luas, menjadikan isyarat ini tidak kelihatan. Oleh itu isyarat daripada homogen B1 medan diperhatikan secara selektif. Peningkatan sensitiviti oleh 1Pengesanan H dalam inv CP-VC mengurangkan sensitiviti yang rendah disebabkan oleh kelimpahan dan volum sampel yang kecil. Secara eksperimen, ia menunjukkan bahawa inv CP-VC mampu memantau a 1H/15N gandingan 2 kHz. CP-VC inv berfungsi sebagai penapisan 1H resonans, memberikan pemerhatian terpilih 1H di sekitar 15N. Ini mengelakkan kerumitan resonans bertindih antara 1Hs. Di samping itu, inv CP-VC membuka jalan untuk menganalisis sampel terhad jisim kurang daripada 1 mg.
Kelewatan ulangan mesti dimasukkan di antara imbasan berturut-turut dalam pengukuran NMR supaya sistem putaran kembali menghampiri keseimbangan terma sebelum memulakan pengukuran NMR. Memandangkan kelewatan pengulangan haruslah tertib T1 masa relaksasi, yang biasanya lebih lama daripada eksperimen yang lain, T1 masa kelonggaran adalah faktor yang mendominasi masa eksperimen dalam NMR. Dalam eksperimen inv CP-VC, kemagnetan bermula dari 1H, oleh itu, 1HT1 ia penting. Walau bagaimanapun, API berat molekul rendah kristal yang baik cenderung mempunyai sangat panjang 1HT1 masa kelonggaran berpuluh-puluh hingga ratusan saat, mengurangkan daya pengeluaran secara mendadak. Pada kadar MAS yang pantas, isunya lebih parah, kerana 1H-1Resapan putaran H ditindas, mengakibatkan lebih lama 1HT1 masa bersantai. Oleh kerana proton NH, yang menjadi minat kami, diasingkan secara spatial dan spektrum daripada 1H yang lain, sangat perlahan 1H-1Resapan putaran H menghalang 1Pemmagnetan H daripada pulih kepada keseimbangan termanya. Ini mengakibatkan lebih perlahan 1HT1 masa kelonggaran proton NH berbanding dengan yang lain 1Hs. Untuk mengatasi kesukaran ini, kami telah menggunakan jujukan penggabungan semula frekuensi radio (RFDR) pada 1H, semasa kelewatan pengulangan [15]. Ini meningkatkan 1H-1Resapan putaran H, membawa kemagnetan daripada mengendur dengan pantas 1H kepada NH proton. Memandangkan mekanisme kelonggaran proton yang mengendur dengan pantas digunakan beberapa kali, bukan sahaja proton NH tetapi juga kepekaan keseluruhan juga boleh dipertingkatkan. Kami juga mengoptimumkan fasa berbasikal dalam urutan RFDR yang sesuai untuk 1H pencampuran [16, 17].
Kesimpulan
Kedua-dua TEM dan NMR terus dipertingkatkan walaupun pada hakikatnya ia telah diperkenalkan lebih 70 tahun yang lalu. Kamera sensitiviti tinggi yang diperkenalkan baru-baru ini dalam TEM membuka jalan kepada aplikasi baru dalam cryo TEM untuk menentukan struktur protein menggunakan pendekatan analisis zarah tunggal. Peningkatan sensitiviti adalah bermanfaat untuk bukan sahaja protein tetapi juga sampel sensitif rasuk termasuk API berat molekul rendah. Satu lagi letusan besar diperhatikan dalam teknologi MAS pantas ssNMR, yang membolehkan 1H NMR walaupun pepejal tegar. Perkembangan baharu ini bukan sahaja menyediakan maklumat baharu yang sebelum ini tidak boleh diakses tetapi juga meningkatkan daya pengeluaran, yang merupakan salah satu faktor utama untuk penyebaran luas penggunaan. Di samping itu, adalah penting untuk menggunakan beberapa kaedah analisis berbeza yang saling melengkapi. Gabungan yang betul memberikan maklumat berguna dengan daya pemprosesan yang tinggi. Sesungguhnya, JEOL Ltd. juga mendorong strategi YOKOGUSHI untuk menawarkan kombinasi peralatan yang menarik.
Dalam artikel ini, kami menggabungkan XRD, ED dan ssNMR untuk menyelesaikan masalah struktur dalam sains farmaseutikal termasuk polimorf kristal dan masalah garam/kokristal/kontinum. Untuk isu polimorf kristal, kami menunjukkan pendekatan gabungan ED dan 1H cepat MAS NMR untuk mengenal pasti bentuk kristal, sebagai kaedah pelengkap kepada PXRD dan 13C CPMAS. ED membolehkan penentuan bentuk kristal daripada kristal tunggal bersaiz nano kepada mikro. Selain itu, 1H cepat MAS boleh membezakan bentuk kristal dengan konformasi molekul yang serupa. Di samping itu, MAS pantas membolehkan 1H/14N korelasi, memberikan kedudukan proton dekat dengan nukleus nitrogen. Untuk mengenal pasti garam/kokristal/kontinum, pendekatan gabungan SCXRD dan ssNMR pada kadar MAS yang sangat pantas diperkenalkan. Walaupun SCXRD menyediakan struktur molekul global, ia gagal menentukan kedudukan proton yang penting untuk isu garam/kokristal/kontinum. Sebaliknya, ssNMR memberikan ketepatan 1H-15N jarak, walaupun struktur global hampir tidak diperolehi. Pendekatan ini memberikan pemahaman yang lancar tentang struktur molekul daripada struktur kristal kepada ikatan hidrogen untuk menjawab isu garam/kokristal/kontinum. Pembentukan kontinum dalam sistem berbilang komponen dengan ΔpKa > 3 menyerlahkan kepentingan penyiasatan menyeluruh menggunakan SCXRD dan ssNMR.
Penghargaan
Semua kerja yang dibentangkan di sini telah dilakukan bersama ahli kumpulan kami di makmal kerjasama RIKEN CLST-JEOL. Saya ingin mengucapkan terima kasih kepada semua ahli kumpulan dan kepada rakan sekerja kami di JEOL Ltd. dan JEOL RESONANCE Inc. atas sokongan yang hebat. Saya juga ingin mengucapkan terima kasih kepada rakan-rakan kami di institut sains India di Bangalore, makmal AMES, Universiti Kyoto, Universiti Pertanian dan Teknologi Tokyo, Universiti Warwick, Universiti Lille, Universiti Michigan atas kerjasama yang membuahkan hasil.
Rujukan
- RK Harris, J. Pharm. Pharmacol. 59 (2007) 225–239.
- M. Geppi, G. Mollica, S. Borsacchi, CA Veracini, Appl. Spectrosc. Rev. 43 (2008) 202–302.
- FG Vogt, Med masa depan. Kimia. 2 (2010) 915–921.
- Y. Nishiyama*, Sampel Sudut Ajaib Pantas Berputar NMR Keadaan Pepejal pada 60-100 kHz untuk Sampel Kelimpahan Semulajadi, Resonans Magnet Nuklear Keadaan Pepejal, 78 (2016) 24-36. DOI: 10. 1016/j. ssnmr. 2016. 06. 002.
- Y. Nishiyama*, NMR keadaan pepejal di bawah kadar MAS ultrapantas 40 ‒ 120 kHz, dalam Pendekatan Eksperimen Spektroskopi NMR ‐ Metodologi dan Aplikasi untuk Sains Hayat dan Sains Bahan, Springer (2017).
- T. Kobayashi, Y. Nishiyama*, M. Pruski*, spektroskopi korelasi Heteronuklear dengan pengesanan songsang, dalam Kaedah Moden dalam NMR Keadaan Pepejal: Panduan Pengamal, Royal Society of Chemistry (2018).
- T. Oikawa, M. Okumura, T. Kimura, Y. Nishiyama*, NMR keadaan pepejal memenuhi pembelauan elektron: Penentuan polimorf hablur bagi sampel mikrohablur rganik kecil, Acta Cryst. C102 (2017) 219‒228. DOI: 10. 1107/S2053229617003084.
- Y. Nishiyama*, Y. Endo, T. Nemoto, H. Utsumi, K. Yamauchi,
Adakah anda seorang profesional perubatan atau kakitangan yang terlibat dalam penjagaan perubatan?
Tidak
Sila diingatkan bahawa halaman ini tidak bertujuan untuk memberikan maklumat tentang produk kepada orang ramai.